Utilisation de modèles de diffusion d’apprentissage profond pour débruiter les données de séquençage d’ARN à faible couverture
Bien que le séquençage de l’ARN (RNA-seq) permette d’obtenir une compréhension approfondie de la biologie humaine, le nombre d’échantillons pouvant être séquencés demeure limité par les coûts associés au séquençage. Il est possible de réduire le coût par échantillon en diminuant la profondeur de séquençage, mais cela entraîne une baisse de la qualité des données. De plus, les méthodes existantes visant à améliorer artificiellement la qualité des données, telles que les approches d’imputation utilisées en RNA-seq unicellulaire, ne sont pas adaptées au débruitage des données de RNA-seq conventionnel (bulk RNA-seq).
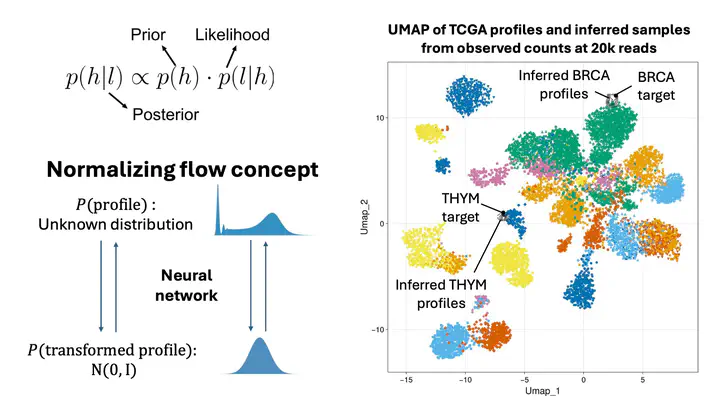
Des réseaux neuronaux de débruitage de type un-à-un ont été développés et permettent de récupérer une grande partie de l’information perdue lors d’un séquençage à faible profondeur. Toutefois, ces modèles ne tiennent pas compte de l’incertitude et du bruit associés à la réduction de la profondeur de séquençage. Nous explorons donc des modèles d’inférence probabiliste, notamment l’inférence bayésienne et l’inférence variationnelle, en tirant parti des normalizing flows et des copules, afin de prédire la distribution des profils transcriptomiques réels susceptibles d’avoir généré les comptages observés à faible profondeur.
Ce nouveau paradigme pourrait contribuer à l’établissement de nouveaux standards de séquençage en RNA-seq dans divers contextes expérimentaux, analytiques ou hospitaliers, permettant à la fois de réduire les coûts et de générer de grandes quantités de données.