Amélioration de la spectrométrie de masse peptidique par l’apprentissage profond
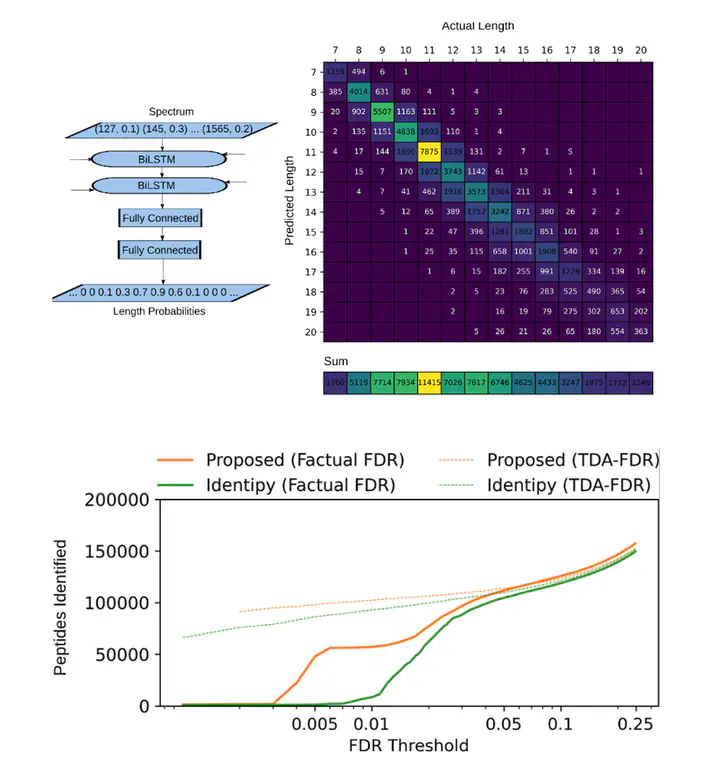
La spectrométrie de masse est l’outil le plus prometteur pour la recherche en protéomique. Cependant, les logiciels d’analyse de spectrométrie de masse à la pointe de la technologie laissent beaucoup à désirer. Les deux principaux paradigmes utilisés sont le séquençage de novo, qui tente d’identifier un peptide simplement à partir de son spectre, et les méthodes de recherche de base de données, qui nécessitent des bases de données bien conçues pour fonctionner. Celles qui sont non spécifiques entraîneront trop de faux positifs et de faux négatifs. Celles qui sont trop grosses entraîneront un décalage dans les algorithmes actuels et les séquences seront incorrectes et des bases de données trop petites ne permettront pas d’obtenir des correspondances sous un seuil de fausses découvertes approprié. De plus, des méthodes axées sur la base de données ne fonctionnent pas correctement lorsqu’aucune base de données n’est disponible, comme c’est le cas pour les peptides associés au CHM-I.
Dans le projet de Jérémie Zumer, l’apprentissage profond est utilisé pour combler les failles de ces méthodes en extrayant des traits jamais disponibles auparavant dans les spectres de masse de manière de novo. Ceci est ensuite utilisé dans un pipeline classique de recherche de base de données avec une fonction de coût ajustée qui prend en compte les traits ajoutés. Le résultat est une amélioration considérable du taux d’identification des peptides sur des ensembles de données tels que dans ProteomeTools : près de 700 % à un seuil de fausses découvertes de 1 % par rapport au logiciel IdentiPy.